
肾上腺皮质癌(adrenocortical carcinoma,ACC)是一种罕见的高度侵袭性的恶性内分泌肿瘤。其估计发病率为0.7×106~2.0×106例/年,1~4岁和40~50岁发病多见,女性患病率较高。ACC可能为有功能的(指具有激素分泌功能),引起库欣综合征和/或男性化;也可能为无功能的,表现为上腹部肿块或者仅偶然发现。该病预后差,5年总生存率仅15%~44%。肿瘤分期是ACC的关键预后因素,Ⅰ期患者的预期5年生存率为80%,而Ⅳ期患者的5年生存率为13%。约2/3的局部疾病患者复发,需要全身化疗为主的综合治疗。
ACC兼具恶性肿瘤与内分泌肿瘤的特点,要关注其侵袭性的生物学行为,还要明确其激素分泌特点;定位与定性的影像学及同位素检查,涉及到多个系统的鉴别诊断;手术治疗与非手术治疗还存在许多争议的方面;其随访不仅要完善内分泌检查,而且要定期复查相关激素、肝肾功能和影像学检查(图1)。
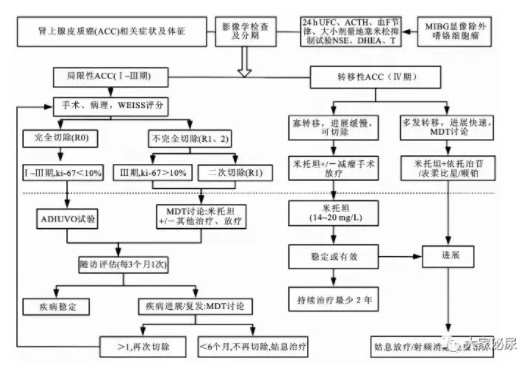
图1 ACC推荐的诊治流程
发病机制
ACC患者中最常见的是胰岛素样生长因子2(IGF2)过度表达和Wnt/β-连环蛋白通路持续激活。IGF2过表达与11p15上的表观遗传印迹发生修饰后的等位基因复制有关。β-连环蛋白的激活与ACC患者的总体生存率下降有关。类固醇因子(SF1)能够促进肾上腺皮质细胞增殖,与ACC患者的不良预后相关。儿童ACC患者中大约有50%~80%发生tp53肿瘤抑癌基因胚系突变(R337H),在巴西南部患病率高达0.27%。Li-Fraumeni综合征的患者最终容易进展成为各种恶性病变,发生tp53突变的肾上腺肿瘤和侵袭性表型相关。有3.2%的ACC患者错配修复基因(mismatch repair, MMR)发生种系突变,可导致Lynch综合征。bub1和pink1基因联合检测有助于对ACC进行亚组分型及预后分析。其他可能的通路有Notch信号通路及肾上腺皮质microRNA谱发生修饰,血清高浓度的miR4835p可能预后不良。此外,CpG岛甲基化可能会引起抑癌基因表达受抑制,转录基因组的数据聚类分析可以鉴别出不同预后的患者。大多数散发病例病因不明。
临床表现及诊断
ACC患者累及肾上腺皮质的球状带、束状带和网状带,肿瘤较大也会挤压肾上腺髓质受累,其主要临床表现为肥胖(91.4%)、库欣综合征体貌(87.1%)、疲乏(81.7%)、女性月经异常(84.3%)、下腹部及大腿根部皮肤紫纹(69.9%)、骨质疏松(57%)。部分ACC患者起病急骤,有腹部包块,伴发热、性激素过量分泌、女性眉毛浓黑、月经不规律或消失、性器官改变等,有典型的皮质醇增多症表现,如满月脸、水牛背、向心性肥胖、毳毛增多、痤疮、宽大紫纹和皮肤易青肿等。疑似ACC的患者都应进行仔细评估,包括病史、临床症状和肾上腺皮质激素自主过量的体征。详细的激素检查,包括血清皮质醇、促肾上腺皮质激素(adreno-cortico-tropic-hormone, ACTH)、尿游离皮质醇(urinary free cortisol, UFC)、17-羟孕酮、脱氢表雄酮、睾酮、雌二醇的测定及大小剂量地塞米松抑制试验。此外,必须排除嗜铬细胞瘤,有无典型的头晕、心悸、冷汗等临床症状、血尿儿茶酚胺水平和间碘苄胍(metaiodoenzylguanidine, MIBG)显像。另外要进行基因检测,确定有无Lynch综合征、Li-Fraumeni综合征及Beck-W综合征等。其他基因如tp53、sf1、igf2、dl7、bub1和pink1,微卫星灶不稳定(microsatellite instability, MSI)、错配基因修复(DNA mismatch repair, MMR)对诊断和治疗也会带来帮助。
影像学检查评估方面,>4 cm的不规则边缘或内部异质的功能性肿瘤应考虑ACC,如怀疑淋巴结或肝转移需行增强电子计算机断层扫描(computed tomography, CT)评估。平扫CT把10 Hu作为区分良性和恶性肾上腺肿瘤的界限,增强的CT在15 min时洗脱值大于60%,肿瘤良性可能性大,核磁共振成像(magnetic resonance imaging, MRI)比CT扫描更清楚地显示局部侵犯和下腔静脉受累情况。无论是CT还是MRI扫描,使用肾上腺区薄扫确定大小、异质性、脂质含量(MRI检查)、对比度清除(CT检查)和边缘特性。当原发肿瘤大于4 cm并且怀疑有癌症时评估转移性疾病和局部入侵时,推荐胸腹盆CT或MRI。联合内分泌功能检查,断层CT可使大多数ACC患者得到正确的诊断。ACC患者的肾上腺肿瘤体积多>11 cm,而大多数腺瘤患者肿瘤体积<5 cm。肾上腺肿瘤检出率呈上升趋势,因此当肾上腺肿瘤直径在3~10 cm时,需慎重诊断。MRI成像技术可以达到同样的准确性,超声检查在伴有肝转移方面有一定的作用。ACC患者都具有高水平的18-氟脱氧葡萄糖摄取,用美托咪酯作为示踪剂进行正电子发射计算机断层显像(positron emission tomographycomputed tomography, PET- CT)检查,美托咪酯可以与肾上腺皮质上的CYP11B酶特异性结合,CYP11B酶是催化皮质醇合成通路过程中的最后一步,这种方法特异性很高。ACC患者需行胸部CT明确有无肺转移,而脑部和骨扫描仅在出现相应的可疑症状时进行。建议不要在疑似患者中行肾上腺活检,除非患者为转移性疾病无法进行手术,需要病理诊断以进行下一步的肿瘤治疗。
ACC主要与嗜铬细胞瘤、肾上腺转移瘤、淋巴瘤及肾上腺区间叶组织来源肿瘤(畸胎瘤、平滑肌瘤、骨肉瘤、淋巴管囊肿等)进行鉴别诊断。较大肿瘤且不适合手术者,需穿刺活检明确病理诊断,行药物治疗或其他非手术治疗。
专家共识推荐:ACC的相关激素应尽量在同一家医疗机构进行检测,同时行影像学检查和核素扫描检查,方便观察并发症及预后情况,减少人为肿瘤的误诊和漏诊。为降低放射暴露,随访可使用MRI检查。
病理与分级、分期
病理诊断要明确病理组织是来源于肾上腺组织,是否表达SF1;另外要鉴别肿瘤的良恶性。目前韦斯(WEISS)评分仍然是用于鉴别肾上腺肿瘤良恶性最好的方法。WEISS评分对肾上腺皮质良、恶肿瘤的9项组织学鉴别标准:核异型大小;核分裂指数≥5/50 HPF;不典型核分裂;透明细胞占全部细胞≤25%;肿瘤细胞呈弥漫性分布;肿瘤坏死;静脉侵犯;窦状样结构浸润;包膜浸润。该系统将9个组织学标准各赋值1分,分数大于3分则为恶性。其中核分裂数目、病理性核分裂象、血管或包膜侵犯以及坏死等是典型的病理组织学恶性指标。而核增殖指数ki-67是重要的预后指标,可以指导治疗。
美国癌症联合委员会(American Joint Committee on Cancer,AJCC)肾上腺皮质癌TNM分期系统(2017年第8版)见表1。TX原发肿瘤不能评估;T1肿瘤证据≤最大尺寸5cm,无肾上腺外侵犯;T2肿瘤>5cm,无肾上腺外侵犯;T3肿瘤,有局部侵犯但未侵犯邻近器官;T4肿瘤,任何大小的肿瘤侵犯邻近器官(肾、膈、胰腺、脾脏或肝脏)或大血管(肾静脉或腔静脉)。NX区域淋巴结无法评估;N0无区域淋巴结转移;N1区域淋巴结转移。M0无远处转移;M1远处转移。G指组织学分级:LG低分级(≤每50 HPF有20个有丝分裂);HG高等级(>20个有丝分裂/50 HPF);TP53或CTNNB突变。
推荐使用ENSAT的肿瘤分期,1期:肿瘤体积≤5cm;2期:肿瘤体积>5cm;3期:肿瘤向周围组织浸润,发生区域淋巴结转移,或者腔静脉/肾静脉有瘤栓形成;4期:指肿瘤发生远隔部位转移。这种分期能够区分不同患者的预后,各个分期5年独立生存期分别为81%、61%、50%和13%。一些分子标志物如基质金属蛋白酶2、葡萄糖转运子1(glucose transporter,GLUT1)、SF1、BUB1和PINK1可能在将来对于分期会有帮助。
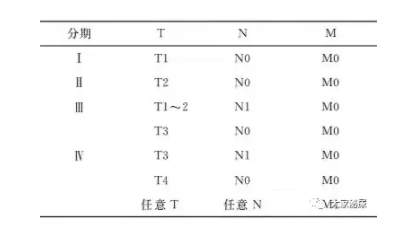
表1 肾上腺皮质癌的临床分期

